Early this week, I was re-organising my bookmarks in the browser and I came across this post by Heng Li published more than 6 years ago. It reminded me one of the problem I faced ~10 years ago when I first started using the GRCh38/hg38 version of the human reference genome for RNA-seq analysis. Let’s have a look what the problem was.
Now if we go to the UCSC genome browser to download the reference genome data, we see that there are a couple of options:
Let’s get the Analysis Set for ease of use since we are doing NGS. By the way, I don’t think this “Analysis Set” thing existed 10 years ago when I encountered the problem. In the download link, we see there are two versions:
.
├── hg38.analysisSet.chroms.tar.gz
└── hg38.fullAnalysisSet.chroms.tar.gz
If we look at the content, we will realise that the following sequences are in both versions:
- Chromosomes
- These are the sequences from regular reference chromosomes
- They are
chr{1..22}.fa,chrX.fa,chrY.faandchrM.fa - In addition,
chrEBV.fais also present as a “decoy” sequence to improve the accuracy of mapping
- Unlocalised scaffolds
- These are the sequences that we know associated with a particular chromosome, but the exact location and orientation are unknown.
- They are named in the format of
chr{chromosome number or name}_{sequence_accession}v{sequence_version}_random - There are a total of 42 of them, e.g.
chr1_KI270706v1_random.fa
- Unplaced scaffolds
- These are the sequences are not associated with any chromosomes
- They are named in the format of
chrUn_{sequence_accession}v{sequence_version} - There are a total of 127 of them, e.g.
chrUn_KI270302v1.fa
Those three types of sequences make up what we called the primary assembly version of a genome. We can think hg38.analysisSet.chroms.tar.gz as the primary assembly of the hg38 version of the human genome. On top of those, there are some extra sequences from hg38.fullAnalysisSet.chroms.tar.gz, they are:
- Alternate loci scaffolds
- These are the alternative haplotypes that we know are present in the population
- They are extremely similar to loci in the primary assembly
- They are named in the format of
chr{chromosome number or name}_{sequence_accession}v{sequence_version}_alt - There are a total of 261 of them, e.g.
chr15_KI270905v1_alt.fa
It is those _alt sequences that cause the problem. Let’s use the sample ERR3153919 from ENA for the demonstration. It is the RNA-seq experiment from the H1 human embryonic stem cell (hESC) line. I’m using STAR v2.7.11a to quantify the gene expression.
To start with the process, we make new directories to hold the reference genome data and raw RNA-seq data (FASTQ files). From here, we simply call the fullAnalysisSet reference genome “full set”, and the analysisSet “no alt”. In addition, we also need the gene annotation file, and we choose to use the RefSeq annotation:
# make three directories to hold ref and actual data
mkdir full_set no_alt fastq gtf
# download the reference genomes and raw data
wget -c 'https://hgdownload.soe.ucsc.edu/goldenPath/hg38/bigZips/analysisSet/hg38.analysisSet.chroms.tar.gz' \
-O no_alt/hg38.analysisSet.chroms.tar.gz
wget -c 'https://hgdownload.soe.ucsc.edu/goldenPath/hg38/bigZips/analysisSet/hg38.fullAnalysisSet.chroms.tar.gz' \
-O full_set/hg38.fullAnalysisSet.chroms.tar.gz
wget -c 'https://hgdownload.soe.ucsc.edu/goldenPath/hg38/bigZips/genes/hg38.ncbiRefSeq.gtf.gz' \
-O gtf/hg38.ncbiRefSeq.gtf.gz
wget -c 'ftp://ftp.sra.ebi.ac.uk/vol1/fastq/ERR315/ERR315391/ERR315391_1.fastq.gz'
-O fastq/ERR315391_1.fastq.gz
wget -c 'ftp://ftp.sra.ebi.ac.uk/vol1/fastq/ERR315/ERR315391/ERR315391_2.fastq.gz'
-O fastq/ERR315391_2.fastq.gz
# unzip the gtf file
gunzip gtf/hg38.ncbiRefSeq.gtf.gz
# extract reference fasta files
tar zxf no_alt/hg38.analysisSet.chroms.tar.gz -C no_alt/
tar zxf full_set/hg38.fullAnalysisSet.chroms.tar.gz -C full_set/
After that, our current directory looks like this:
.
├── fastq
│ ├── ERR3153919_1.fastq.gz
│ └── ERR3153919_2.fastq.gz
├── full_set
│ ├── hg38.fullAnalysisSet.chroms
│ │ ├── chr10.fa
│ │ ├── ........
│ │ └── chrY_KI270740v1_random.fa
│ └── hg38.fullAnalysisSet.chroms.tar.gz
├── gtf
│ └── hg38.ncbiRefSeq.gtf
└── no_alt
├── hg38.analysisSet.chroms
│ ├── chr10.fa
│ ├── ........
│ └── chrY_KI270740v1_random.fa
└── hg38.analysisSet.chroms.tar.gz
Now, let’s make star index:
# for full_set
STAR --runThreadN 20 \
--runMode genomeGenerate \
--genomeDir full_set/star_index/ \
--genomeFastaFiles full_set/hg38.fullAnalysisSet.chroms/*.fa \
--sjdbGTFfile gtf/hg38.ncbiRefSeq.gtf
# for no_alt
STAR --runThreadN 20 \
--runMode genomeGenerate \
--genomeDir no_alt/star_index/ \
--genomeFastaFiles no_alt/hg38.analysisSet.chroms/*.fa \
--sjdbGTFfile gtf/hg38.ncbiRefSeq.gtf
Then we perform the actual alignment and quantification:
# for full_set
STAR --runThreadN 20 \
--genomeDir full_set/star_index \
--readFilesIn fastq/ERR3153919_1.fastq.gz fastq/ERR3153919_2.fastq.gz \
--readFilesCommand zcat \
--quantMode GeneCounts \
--outFileNamePrefix ./star_outs/full_set/
# for no_alt
STAR --runThreadN 20 \
--genomeDir no_alt/star_index \
--readFilesIn fastq/ERR3153919_1.fastq.gz fastq/ERR3153919_2.fastq.gz \
--readFilesCommand zcat \
--quantMode GeneCounts \
--outFileNamePrefix ./star_outs/no_alt/
# index and sort the output sam file for future use
samtools view -u star_outs/full_set/Aligned.out.sam | \
samtools sort - -T tmp -o star_outs/full_set/Aligned.out.sorted.bam
samtools index star_outs/full_set/Aligned.out.sorted.bam
samtools view -u star_outs/no_alt/Aligned.out.sam | \
samtools sort - -T tmp -o star_outs/no_alt/Aligned.out.sorted.bam
samtools index star_outs/no_alt/Aligned.out.sorted.bam
After the above processes are finished, we should have the following files in the star_outs directory:
star_outs/
├── full_set
│ ├── Aligned.out.sam
│ ├── Aligned.out.sorted.bam
│ ├── Aligned.out.sorted.bam.bai
│ ├── Log.final.out
│ ├── Log.out
│ ├── Log.progress.out
│ ├── ReadsPerGene.out.tab
│ └── SJ.out.tab
└── no_alt
├── Aligned.out.sam
├── Aligned.out.sorted.bam
├── Aligned.out.sorted.bam.bai
├── Log.final.out
├── Log.out
├── Log.progress.out
├── ReadsPerGene.out.tab
└── SJ.out.tab
2 directories, 16 files
Now we could create a scatter plot using the 2nd column of ReadsPerGene.out.tab, which is a tab-delimited file containing the read count of each gene in the sample. Plotting full_set vs no_alt in log scale, we have:
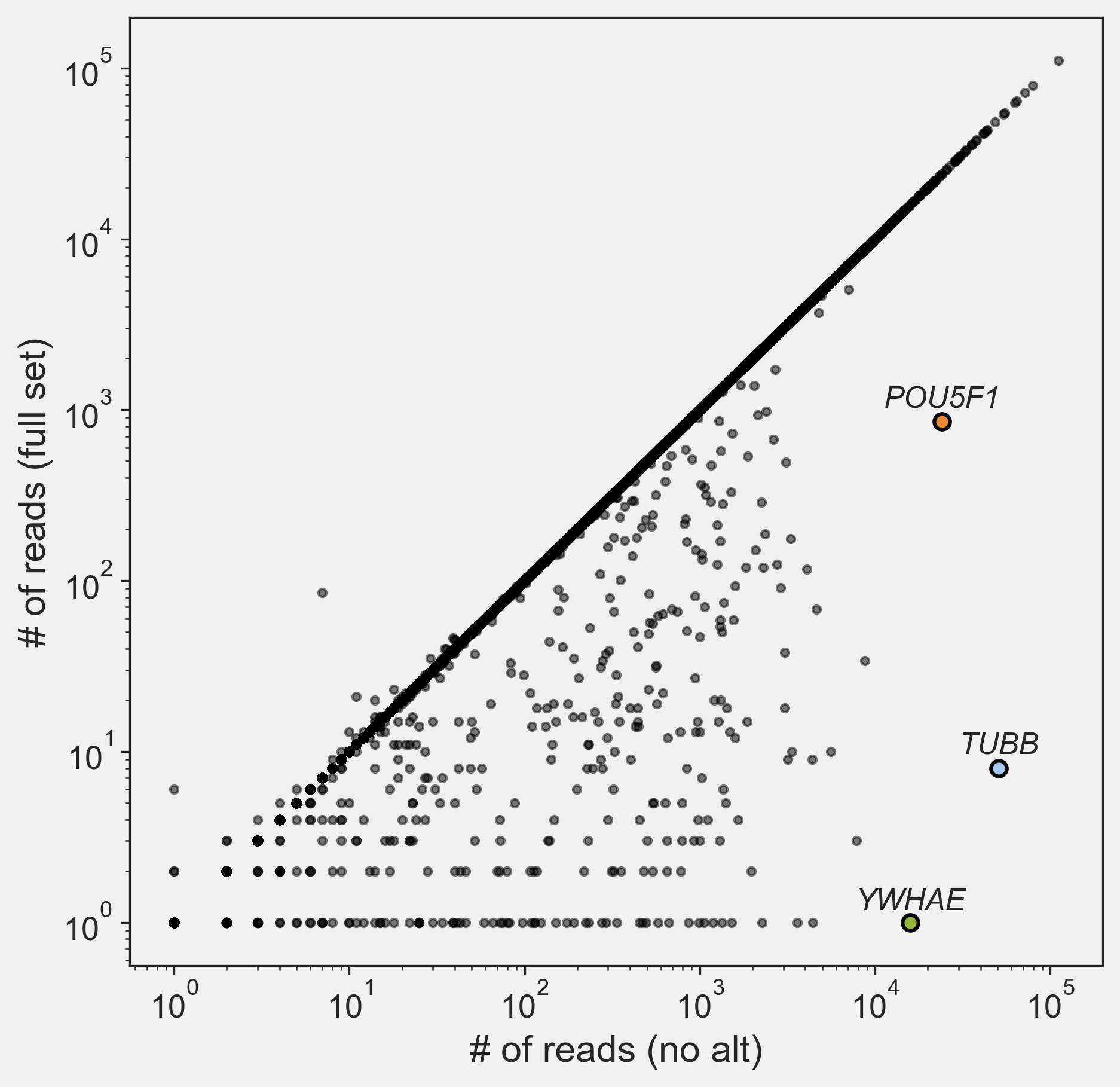
Well … If we look at the scatter, the vast majority of the genes align perfectly on the line $y=x$, which is good. However, 1761 genes are not on the line, most of which have orders of magnitudes more reads in the “no alt” version compared to the “full set”. The three genes at the outermost that are highlighted are OCT4 (POU5F1), beta tubulin (TUBB) and 14-3-3$\varepsilon$ (YWHAE). Let’s use the famous POU5F1 gene as an example. If you work on embryonic stem cells or embryo development, you definitely know this gene. There were 23,992 reads mapped to this gene when using the “no alt” version, but only 851 reads were mapped to this gene when using the “full set” version of the genome. For the exact the same data set, the difference is huge. What is going on here? First, we could have a look at where the gene POU5F1 is located:
grep -w 'gene_name "POU5F1"' gtf/hg38.ncbiRefSeq.gtf
Then we realise that there are multiple records of POU5F1:
chr6 ncbiRefSeq.2022-10-28 transcript 31164337 31167170 . - . gene_id "POU5F1"; transcript_id "NM_001285986.2_7"; gene_name "POU5F1";
chr6 ncbiRefSeq.2022-10-28 transcript 31164337 31167170 . - . gene_id "POU5F1"; transcript_id "NM_203289.6_7"; gene_name "POU5F1";
chr6 ncbiRefSeq.2022-10-28 transcript 31164337 31167170 . - . gene_id "POU5F1"; transcript_id "NM_001173531.3_7"; gene_name "POU5F1";
chr6 ncbiRefSeq.2022-10-28 transcript 31164337 31170682 . - . gene_id "POU5F1"; transcript_id "NM_002701.6_7"; gene_name "POU5F1";
chr6_GL000256v2_alt ncbiRefSeq.2022-10-28 transcript 2467753 2470601 . - . gene_id "POU5F1"; transcript_id "NM_001285986.2"; gene_name "POU5F1";
chr6_GL000256v2_alt ncbiRefSeq.2022-10-28 transcript 2467753 2470601 . - . gene_id "POU5F1"; transcript_id "NM_203289.6"; gene_name "POU5F1";
chr6_GL000256v2_alt ncbiRefSeq.2022-10-28 transcript 2467753 2470601 . - . gene_id "POU5F1"; transcript_id "NM_001173531.3"; gene_name "POU5F1";
chr6_GL000256v2_alt ncbiRefSeq.2022-10-28 transcript 2467753 2474114 . - . gene_id "POU5F1"; transcript_id "NM_002701.6"; gene_name "POU5F1";
chr6_GL000255v2_alt ncbiRefSeq.2022-10-28 transcript 2422370 2425218 . - . gene_id "POU5F1"; transcript_id "NM_001285986.2_2"; gene_name "POU5F1";
chr6_GL000255v2_alt ncbiRefSeq.2022-10-28 transcript 2422370 2425218 . - . gene_id "POU5F1"; transcript_id "NM_203289.6_2"; gene_name "POU5F1";
chr6_GL000255v2_alt ncbiRefSeq.2022-10-28 transcript 2422370 2425218 . - . gene_id "POU5F1"; transcript_id "NM_001173531.3_2"; gene_name "POU5F1";
chr6_GL000255v2_alt ncbiRefSeq.2022-10-28 transcript 2422370 2428732 . - . gene_id "POU5F1"; transcript_id "NM_002701.6_2"; gene_name "POU5F1";
chr6_GL000254v2_alt ncbiRefSeq.2022-10-28 transcript 2508452 2511300 . - . gene_id "POU5F1"; transcript_id "NM_001285986.2_3"; gene_name "POU5F1";
chr6_GL000254v2_alt ncbiRefSeq.2022-10-28 transcript 2508452 2511300 . - . gene_id "POU5F1"; transcript_id "NM_203289.6_3"; gene_name "POU5F1";
chr6_GL000254v2_alt ncbiRefSeq.2022-10-28 transcript 2508452 2511300 . - . gene_id "POU5F1"; transcript_id "NM_001173531.3_3"; gene_name "POU5F1";
chr6_GL000254v2_alt ncbiRefSeq.2022-10-28 transcript 2508452 2514816 . - . gene_id "POU5F1"; transcript_id "NM_002701.6_3"; gene_name "POU5F1";
chr6_GL000253v2_alt ncbiRefSeq.2022-10-28 transcript 2474838 2477686 . - . gene_id "POU5F1"; transcript_id "NM_001285986.2_4"; gene_name "POU5F1";
chr6_GL000253v2_alt ncbiRefSeq.2022-10-28 transcript 2474838 2477686 . - . gene_id "POU5F1"; transcript_id "NM_203289.6_4"; gene_name "POU5F1";
chr6_GL000253v2_alt ncbiRefSeq.2022-10-28 transcript 2474838 2477686 . - . gene_id "POU5F1"; transcript_id "NM_001173531.3_4"; gene_name "POU5F1";
chr6_GL000253v2_alt ncbiRefSeq.2022-10-28 transcript 2474838 2481202 . - . gene_id "POU5F1"; transcript_id "NM_002701.6_4"; gene_name "POU5F1";
chr6_GL000252v2_alt ncbiRefSeq.2022-10-28 transcript 2423653 2426501 . - . gene_id "POU5F1"; transcript_id "NM_001285986.2_5"; gene_name "POU5F1";
chr6_GL000252v2_alt ncbiRefSeq.2022-10-28 transcript 2423653 2426501 . - . gene_id "POU5F1"; transcript_id "NM_203289.6_5"; gene_name "POU5F1";
chr6_GL000252v2_alt ncbiRefSeq.2022-10-28 transcript 2423653 2426501 . - . gene_id "POU5F1"; transcript_id "NM_001173531.3_5"; gene_name "POU5F1";
chr6_GL000252v2_alt ncbiRefSeq.2022-10-28 transcript 2423653 2430016 . - . gene_id "POU5F1"; transcript_id "NM_002701.6_5"; gene_name "POU5F1";
chr6_GL000251v2_alt ncbiRefSeq.2022-10-28 transcript 2646761 2649609 . - . gene_id "POU5F1"; transcript_id "NM_001285986.2_6"; gene_name "POU5F1";
chr6_GL000251v2_alt ncbiRefSeq.2022-10-28 transcript 2646761 2649609 . - . gene_id "POU5F1"; transcript_id "NM_203289.6_6"; gene_name "POU5F1";
chr6_GL000251v2_alt ncbiRefSeq.2022-10-28 transcript 2646761 2649609 . - . gene_id "POU5F1"; transcript_id "NM_001173531.3_6"; gene_name "POU5F1";
chr6_GL000251v2_alt ncbiRefSeq.2022-10-28 transcript 2646761 2653124 . - . gene_id "POU5F1"; transcript_id "NM_002701.6_6"; gene_name "POU5F1";
As we can see, there are multiple transcripts (isoforms) for the same gene POU5F1, but the true record is those from chr6. Other records from chr6_GL00025{1..6}v2_alt are just alternative haplotypes in the population. They have nearly identical sequences to the segment from chr6. Let’s see some reads mapped to POU5F1, starting from the “no alt” version:
$ samtools view star_outs/no_alt/Aligned.out.sorted.bam chr6:31,164,337-31,167,170 | head -5
ERR3153919.15777041 163 chr6 31062785 255 14M217932N60M1S = 31280754 218041 GGCAGGATGACACAGGCAAGAAGCAGCTGGAGAAGGACTTCAACAGCATGAAGAAGTACTGCCAGGTCATCCGCA BBBBBFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFF NH:i:1 HI:i:1 AS:i:114 nM:i:10
ERR3153919.8228362 163 chr6 31062786 255 13M217932N60M2S = 31280842 218124 GCAGGATGACACAGGCAAGAAGCAGCTGGAGAAGGACTTCAACAGCATGAAGAAGTACTGCCAGGTCATCCGCAT BBBBBFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFF NH:i:1 HI:i:1 AS:i:111 nM:i:9
ERR3153919.8525639 419 chr6 31062789 1 10M217932N65M = 31280833 218116 GGATGACACAGGCAAGAAGCAGCTGGAGAAGGACTTCAACAGCATGAAGAAGTACTGCCAGGTCATCCGCATCAT BBBBBFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFF NH:i:3 HI:i:3 AS:i:115 nM:i:10
ERR3153919.13201791 419 chr6 31062789 1 10M217932N65M = 31280837 218116 GGATGACACAGGCAAGAAGCAGCTGGAGAAGGACTTCAACAGCATGAAGAAGTACTGCCAGGTCATCCGCATCAT BB/B/<BFFF/BB//FFFF<////B///<<F</F<FFB<FB/FF<//<//B<<////<B///<FFBFBB/<77<F NH:i:3 HI:i:3 AS:i:111 nM:i:10
ERR3153919.18818531 419 chr6 31062789 1 10M217932N65M = 31280836 218116 GGATGACACAGGCAAGAAGCAGCTGGAGAAGGACTTCAACAGCATGAAGAAGTACTGCCAGGTCATCCGCATCAT BBBBBFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFF NH:i:3 HI:i:3 AS:i:112
Note the first read ERR3153919.15777041 is a good one that has a mapping quality (MAPQ) of 255, indicating it is a uniquely mapped read. Let’s look at where this read would map to in the “full set” version of the genome:
$ samtools view star_outs/full_set/Aligned.out.sorted.bam | grep -w 'ERR3153919.15777041'
ERR3153919.15777041 419 chr6 31062785 0 14M217932N60M1S = 31280754 218041 GGCAGGATGACACAGGCAAGAAGCAGCTGGAGAAGGACTTCAACAGCATGAAGAAGTACTGCCAGGTCATCCGCA BBBBBFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFF NH:i:7 HI:i:7 AS:i:114 nM:i:10
ERR3153919.15777041 339 chr6 31280754 0 72M3S = 31062785 -218041 CTTCAACAGCATGAAGAAGTACTGCCAGGTCATCCGCATCATTGCCCACACTCAGATGCGTCTGCTTCCTCTACG BFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFBBBBB NH:i:7 HI:i:7 AS:i:114 nM:i:10
ERR3153919.15777041 419 chr6_GL000251v2_alt 2545163 0 14M216464N60M1S = 2761664 216573 GGCAGGATGACACAGGCAAGAAGCAGCTGGAGAAGGACTTCAACAGCATGAAGAAGTACTGCCAGGTCATCCGCA BBBBBFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFF NH:i:7 HI:i:4 AS:i:114 nM:i:10
ERR3153919.15777041 339 chr6_GL000251v2_alt 2761664 0 72M3S = 2545163 -216573 CTTCAACAGCATGAAGAAGTACTGCCAGGTCATCCGCATCATTGCCCACACTCAGATGCGTCTGCTTCCTCTACG BFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFBBBBB NH:i:7 HI:i:4 AS:i:114 nM:i:10
ERR3153919.15777041 419 chr6_GL000252v2_alt 2322157 0 14M216300N60M1S = 2538494 216409 GGCAGGATGACACAGGCAAGAAGCAGCTGGAGAAGGACTTCAACAGCATGAAGAAGTACTGCCAGGTCATCCGCA BBBBBFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFF NH:i:7 HI:i:5 AS:i:114 nM:i:10
ERR3153919.15777041 339 chr6_GL000252v2_alt 2538494 0 72M3S = 2322157 -216409 CTTCAACAGCATGAAGAAGTACTGCCAGGTCATCCGCATCATTGCCCACACTCAGATGCGTCTGCTTCCTCTACG BFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFBBBBB NH:i:7 HI:i:5 AS:i:114 nM:i:10
ERR3153919.15777041 419 chr6_GL000253v2_alt 2373292 0 14M216426N60M1S = 2589755 216535 GGCAGGATGACACAGGCAAGAAGCAGCTGGAGAAGGACTTCAACAGCATGAAGAAGTACTGCCAGGTCATCCGCA BBBBBFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFF NH:i:7 HI:i:6 AS:i:114 nM:i:10
ERR3153919.15777041 339 chr6_GL000253v2_alt 2589755 0 72M3S = 2373292 -216535 CTTCAACAGCATGAAGAAGTACTGCCAGGTCATCCGCATCATTGCCCACACTCAGATGCGTCTGCTTCCTCTACG BFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFBBBBB NH:i:7 HI:i:6 AS:i:114 nM:i:10
ERR3153919.15777041 419 chr6_GL000254v2_alt 2406901 0 14M216511N60M1S = 2623449 216620 GGCAGGATGACACAGGCAAGAAGCAGCTGGAGAAGGACTTCAACAGCATGAAGAAGTACTGCCAGGTCATCCGCA BBBBBFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFF NH:i:7 HI:i:1 AS:i:114 nM:i:10
ERR3153919.15777041 339 chr6_GL000254v2_alt 2623449 0 72M3S = 2406901 -216620 CTTCAACAGCATGAAGAAGTACTGCCAGGTCATCCGCATCATTGCCCACACTCAGATGCGTCTGCTTCCTCTACG BFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFBBBBB NH:i:7 HI:i:1 AS:i:114 nM:i:10
ERR3153919.15777041 163 chr6_GL000255v2_alt 2320749 0 14M215351N60M1S = 2536137 215460 GGCAGGATGACACAGGCAAGAAGCAGCTGGAGAAGGACTTCAACAGCATGAAGAAGTACTGCCAGGTCATCCGCA BBBBBFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFF NH:i:7 HI:i:2 AS:i:114 nM:i:10
ERR3153919.15777041 83 chr6_GL000255v2_alt 2536137 0 72M3S = 2320749 -215460 CTTCAACAGCATGAAGAAGTACTGCCAGGTCATCCGCATCATTGCCCACACTCAGATGCGTCTGCTTCCTCTACG BFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFBBBBB NH:i:7 HI:i:2 AS:i:114 nM:i:10
ERR3153919.15777041 419 chr6_GL000256v2_alt 2366233 0 14M216393N60M1S = 2582663 216502 GGCAGGATGACACAGGCAAGAAGCAGCTGGAGAAGGACTTCAACAGCATGAAGAAGTACTGCCAGGTCATCCGCA BBBBBFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFF NH:i:7 HI:i:3 AS:i:114 nM:i:10
ERR3153919.15777041 339 chr6_GL000256v2_alt 2582663 0 72M3S = 2366233 -216502 CTTCAACAGCATGAAGAAGTACTGCCAGGTCATCCGCATCATTGCCCACACTCAGATGCGTCTGCTTCCTCTACG BFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFBBBBB NH:i:7 HI:i:3 AS:i:114 nM:i:10
As we can see, the same read was mapped to 7 locations1 in the “full set” version of the genome. As a result, the mapping quality of this read is 0. One of those alignments was set as the primary alignment, which is the one mapped to chr6_GL000255v2_alt. That is, the pair of reads whose flags are 163 and 83. I think the primary alignment is chosen randomly when all alignments are equally good, but I’m not entirely sure. The rest of the 6 alignments all have the 0x100 flag, that is, not primary alignment. Because of this, the read ERR3153919.15777041 will not be used for quantification. There are many reads like this.
If the “full set” version is used, we would think there might be something wrong with our experiments, because POU5F1 should be highly expressed in any embryonic stem cells. This is actually a good thing so you can investigate what’s going on. However, if we are dealing with some other samples, we may not notice the problem. I’m not sure if the difference of those 1761 genes would affect our major conclusions. The dangerous situation is always that we don’t know we don’t know!
Anyway, this is first reminder in the new year. I tweeted about this, and you can think of this post as a more detailed version of the tweet with code.
P.S. When I finished writing this post, I was wondering why I did not encounter the problem when using hg19 where the _alt chromosomes were also included in the reference. Then I realised I was using the Illumina iGenomes package where those things are removed. Oh well … I guess the take-home message is know your package and read the README carefully.
Note the data is in pair end format, so Read 1 and Read 2 have the same name. We have a total of 14 records, so it is 14/2 = 7 locations. We can also see the optional field
NH:i:7indicated the read is mapped to 7 locations in the genome. ↩︎