In recent years, I noticed one problem about the ATAC-seq/scATAC-seq experiments we did in the lab. Specifically, it is related to the ATAC-seq insert size, or fragment length, plotting. The distribution of the fragment length in an ATAC-seq experiment is an important quality control metric. Since the transposase Tn5 can only go to those nucleosome-free regions, the lengths of the fragments after Tn5 transposition often exhibit a nucleosomal ladder pattern, like shown in this post where I described how to draw the plot by looking at the 9th column of the aligned SAM/BAM file.
However, the fragment length distribution looks like this in all recent experiments we had:
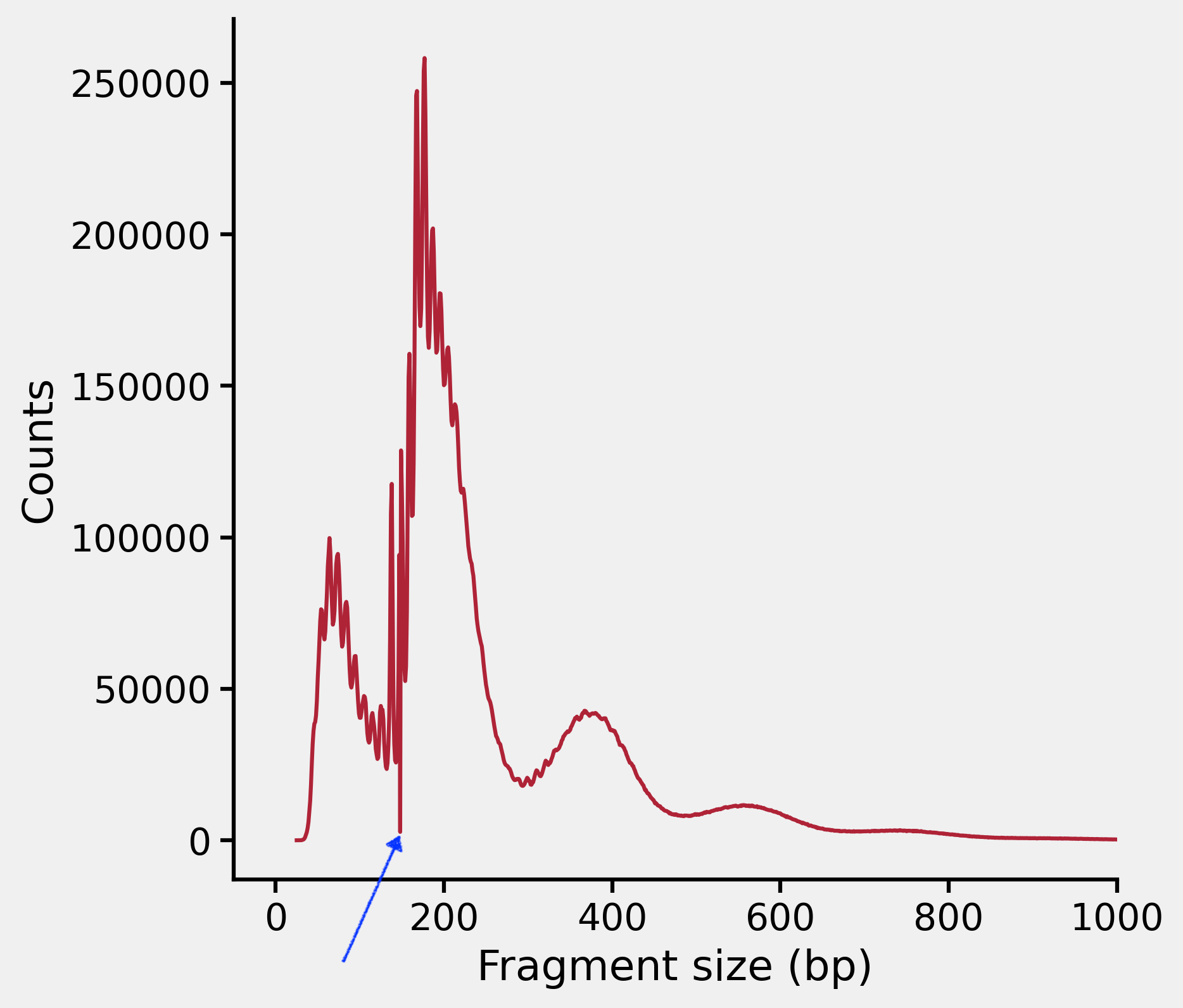
Note the sudden drop of frequency around 150 bp indicated by the blue arrow. This is something unusual.
The ATAC-seq library is the same as the Nextera library since they use the same type of Tn5 for the experiment. The sequencing scheme is like this:
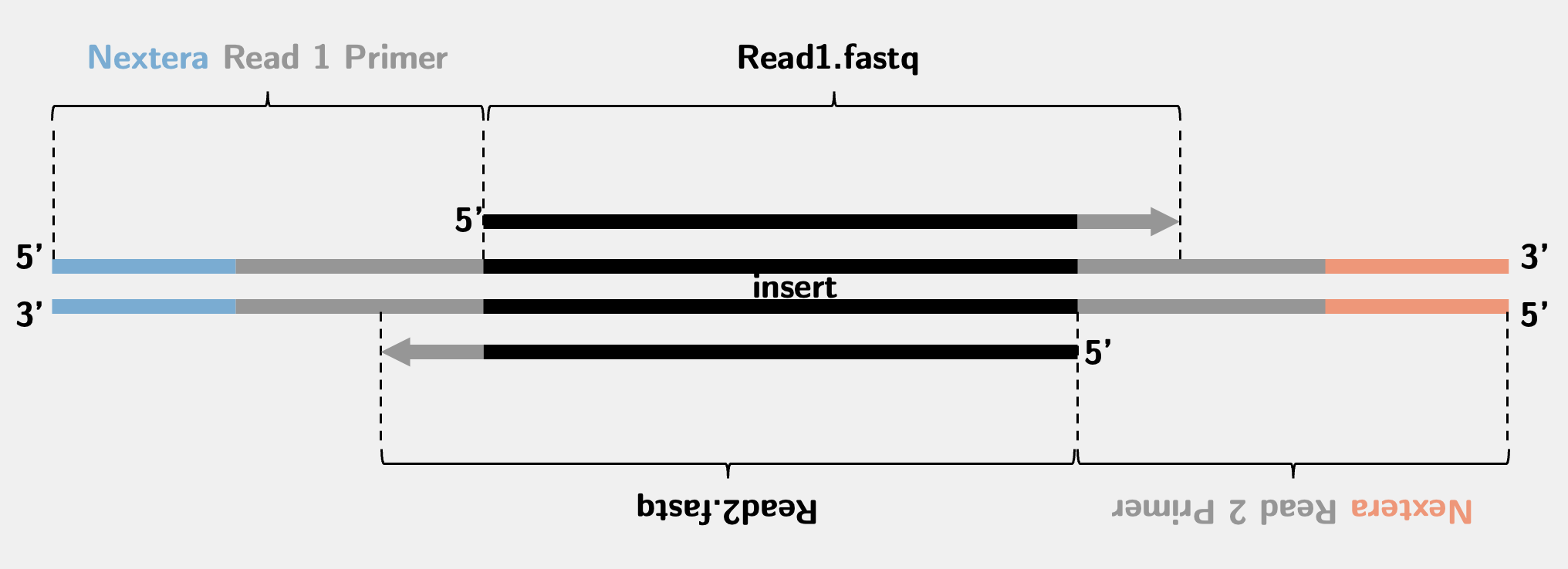
In an ATAC-seq experiment, the vast majority of the fragments, that is the insert between two sequencing primers, are very short. In early days of the sequencing when the read length was quite short (25 - 36 bp), there was no problem if you directly mapped the sequencing reads to the genome. In modern days, people tend to sequence much longer, typically 50 - 150 bp. In this case, many reads will go over the genomic insert and read into the sequencing primer regions, causing a contamination at the 3’ end of the read indicated by the grey arrows in the above picture.
Therefore, in a typical ATAC-seq preprocessing workflow we always trimmed the adapter sequence by cutadapt. The command we use (with v4.5) is as follows:
cutadapt -j 20 -m 25 \
-a CTGTCTCTTATACACATCTCCGAGCCCACGAGAC \
-A CTGTCTCTTATACACATCTGACGCTGCCGACGA \
-o {output.r1} -p {output.r2} \
<r1.fq.gz> <r2.fq.gz> \
1> out.stdout 2> out.stderr
At present I figured out the unusual fragment length distribution was basically caused by something happened during the adapter trimming step performed by cutadapt, because the problem disappeared when I only used the first 36 bp of the reads for the mapping or used fastp for the adapter trimming, as shown below:
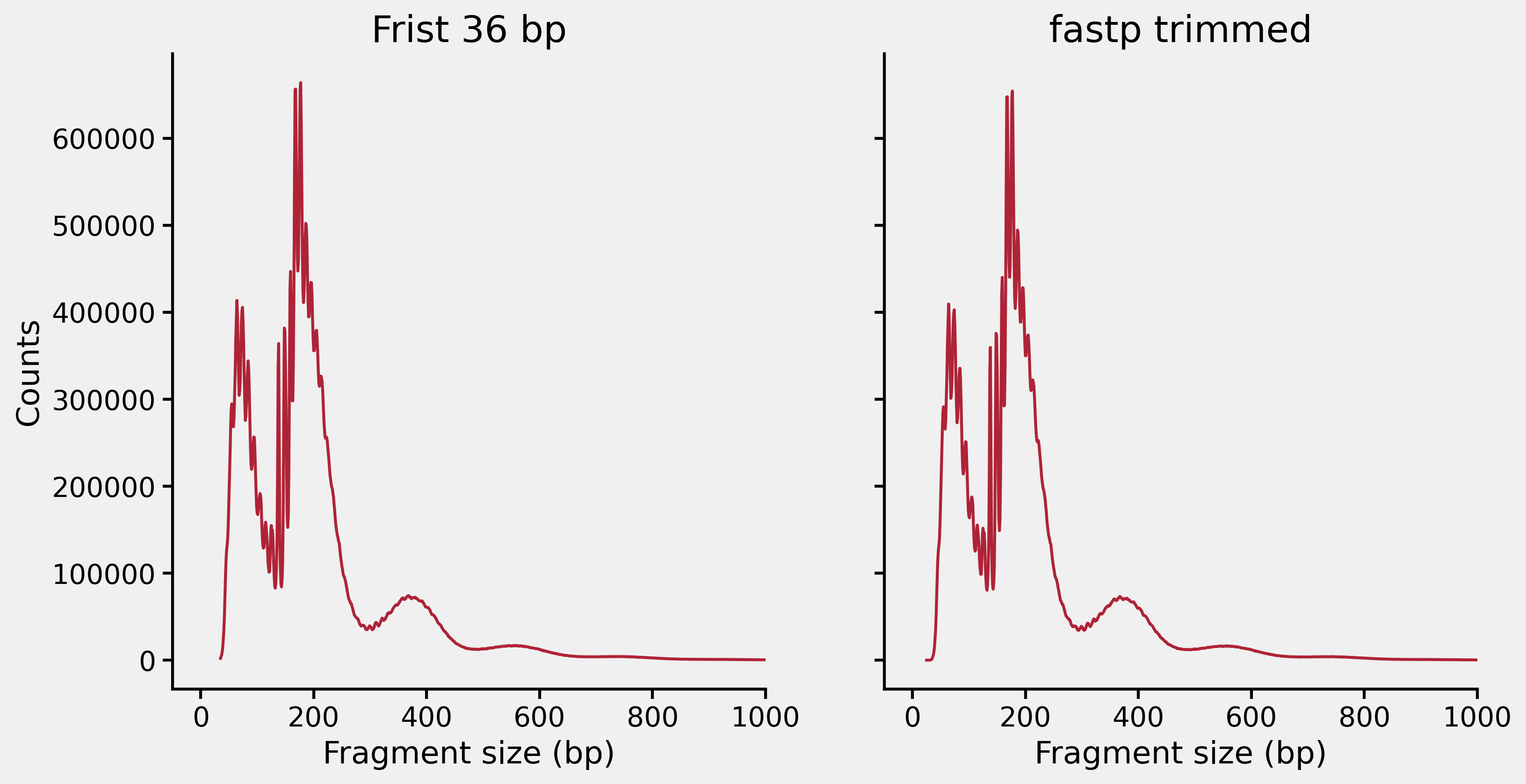
Now I’m documenting how I found out the problem. Let’s use a public ATAC-seq data to demonstrate the point so that you can reproduce the results if you want. I’m using SRR5837342 which is an ATAC-seq from the H1 hESC cell line. The fastq files can be downloaded from this ENA link. Once we have the paired end fastq files, we could start with the easiest:
# simply use the first 36 bp of the reads for the mapping without trimming
# SRR5837342 was using 150 bp PE sequencing, so we passed -3 114 option to bowtie2
bowtie2 -p 20 -X 2000 -3 114 \
--very-sensitive \
-x <hg38_bwt2_idx> \
-1 SRR5837342_1.fastq.gz -2 SRR5837342_2.fastq.gz | \
samtools view -@ 20 -hu - | \
samtools sort - -@ 20 -T tmp -o first36bp_sorted.bam
# now we plot the fragment (isize)
samtools view -F 4 -f 66 -q 30 first36bp_sorted.bam | \ # remove un-mapped reads (-F4), only look at read 1 (-f64) and properly paired reads (-f2), only look at "uniquely" mapped reads (-q30)
sed '/chrM/d' | \ # remove MT reads
awk '{ if($9>0) {print $9} else {print -1*$9} }' | \ # output insert size
sort | uniq -c | sort -b -k2,2n | \ # check distribution of each insert size
awk 'BEGIN{OFS="\t"}{print $2, $1}' > first36bp_f2q30_isize.tsv # format output
Then we can have a look at our typical workflow where cutadapt was used to trim the adapters, and then the same steps were performed:
# adapter removal
cutadapt -j 20 -m 25 \
-a CTGTCTCTTATACACATCTCCGAGCCCACGAGAC \
-A CTGTCTCTTATACACATCTGACGCTGCCGACGA \
-o cutadapt_r1.fq.gz -p cutadapt_r2.fq.gz \
SRR5837342_1.fastq.gz SRR5837342_2.fastq.gz \
1> cutadapt.stdout 2> cutadapt.stderr
# bowtie2 mapping
bowtie2 -p 20 -X 2000 \
--very-sensitive \
-x <hg38_bwt2_idx> \
-1 cutadapt_r1.fq.gz -2 cutadapt_r2.fq.gz | \
samtools view -@ 20 -hu - | \
samtools sort - -@ 20 -T tmp -o cutadapt_sorted.bam
# look at the fragment (isize)
samtools view -F 4 -f 66 -q 30 cutadapt_sorted.bam | \
sed '/chrM/d' | \
awk '{ if($9>0) {print $9} else {print -1*$9} }' | \
sort | uniq -c | sort -b -k2,2n | \
awk 'BEGIN{OFS="\t"}{print $2, $1}' > cutadapt_f2q30_isize.tsv
Then, let’s do the same analysis with fastp as the trimming programme:
# adapter removal
fastp -l 25 -w 16 -x \
--detect_adapter_for_pe \
-i SRR5837342_1.fastq.gz -I SRR5837342_2.fastq.gz \
-o fastp_r1.fq.gz -O fastp_r2.fq.gz
# bowtie2 mapping
bowtie2 -p 20 -X 2000 \
--very-sensitive \
-x <hg38_bwt2_idx> \
-1 fastp_r1.fq.gz -2 fastp_r2.fq.gz | \
samtools view -@ 20 -hu - | \
samtools sort - -@ 20 -T tmp -o fastp_sorted.bam
# look at the fragment (isize)
samtools view -F 4 -f 66 -q 30 fastp_sorted.bam | \
sed '/chrM/d' | \
awk '{ if($9>0) {print $9} else {print -1*$9} }' | \
sort | uniq -c | sort -b -k2,2n | \
awk 'BEGIN{OFS="\t"}{print $2, $1}' > fastp_f2q30_isize.tsv
Now we could look at the insert distributions from those three different tsv files, which is shown below:
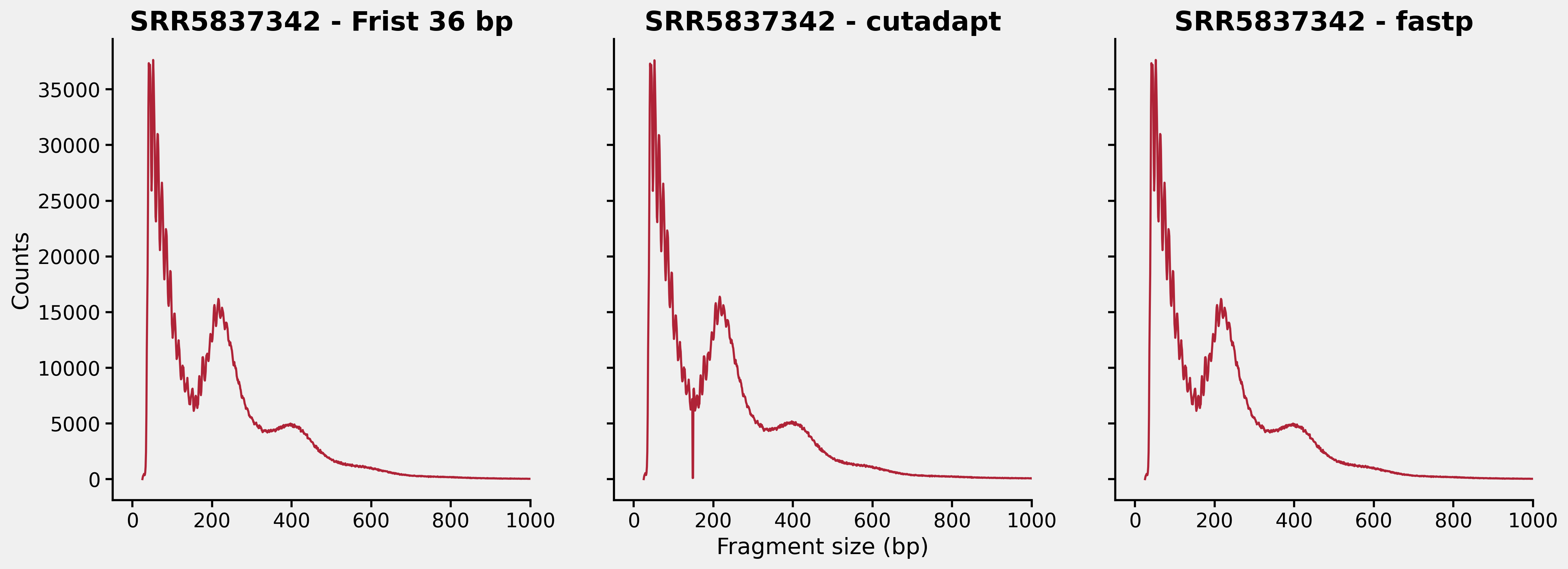
Apparently, there are something wrong with our cutadapt workflow. First, let’s have a look what fragment lengths are kind of “depleted” in the cutadapt_f2q30_isize.tsv. This is the content of the file around lines 122 - 127:
| |
Oh, it turned out that very few mapped fragments had a length of 148 or 149 bp, which is really weird. Apparently, there are many fragments with length 148 or 149 bp in the fastp workflow. We could extract all fragments with length of 148 or 149 bp from the fastp workflow and look at how they are treated in the cutadapt workflow. They might be filtered out by any of the -F4 -f66 -q30 flags in the preprocessing. I think it is better to go back to the original fastq files to have a look. To this end, we extract all Read IDs with length of 148 or 149 bp from fastp_sorted.bam, and get the fastq reads based on the Read IDs:
# extract isize==148 or 149, keep Read ID and isize which might be useful
samtools view -F 4 -f 66 -q 30 fastp_sorted.bam | \
sed '/chrM/d' | \
awk '($9==148)||($9==-148)||($9==149)||($9==-149)' | \
cut -f 1,9 | sort -k1,1 > fastp_f2q30_read_isize_148_149.tsv
# use seqtk to get fastq reads
## get original reads
seqtk subseq SRR5837342_1.fastq.gz <(cut -f 1 fastp_f2q30_read_isize_148_149.tsv) | \
gzip > SRR5837342_isize148_149_r1.fq.gz
seqtk subseq SRR5837342_2.fastq.gz <(cut -f 1 fastp_f2q30_read_isize_148_149.tsv) | \
gzip > SRR5837342_isize148_149_r2.fq.gz
## get the same set of reads from cutadapt reads
seqtk subseq cutadapt_r1.fq.gz <(cut -f 1 fastp_f2q30_read_isize_148_149.tsv) | \
gzip > cutadapt_isize148_149_r1.fq.gz
seqtk subseq cutadapt_r2.fq.gz <(cut -f 1 fastp_f2q30_read_isize_148_149.tsv) | \
gzip > cutadapt_isize148_149_r2.fq.gz
## get the same set of reads from fastp reads
seqtk subseq fastp_r1.fq.gz <(cut -f 1 fastp_f2q30_read_isize_148_149.tsv) | \
gzip > fastp_isize148_149_r1.fq.gz
seqtk subseq cutadapt_r2.fq.gz <(cut -f 1 fastp_f2q30_read_isize_148_149.tsv) | \
gzip > fastp_isize148_149_r2.fq.gz
Now we are ready to manually look at some reads. This is the first ten lines of fastp_f2q30_read_isize_148_149.tsv:
SRR5837342.10000218 -148
SRR5837342.10000523 -149
SRR5837342.10000984 -149
SRR5837342.100014 149
SRR5837342.10001497 -149
SRR5837342.10001822 -149
SRR5837342.10002543 148
SRR5837342.10002643 149
SRR5837342.10003834 -149
SRR5837342.10003929 148
Now we can have a look at the read SRR5837342.10000218. First, let’s look at it from the original fastq files:
$ zcat SRR5837342_isize148_149_r1.fq.gz | grep -A 3 -w 'SRR5837342.10000218'
@SRR5837342.10000218 10000218/1
CGTGACGGCCTCTGCGTGCCTACGTGCGCGCCCCCGGCCGGGAGCTTCTAGCGCTTGTCCCGCCCTCCAGGCCTGCCTAGCGCGGCGCGGGGCTGCTGCGCCTGCGCGCTCGCGGCTTGCCGCGGCCCGGAGCGCAGCCCAGCTGTCACT
+
AAFFFJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJFJJJJJJJJJJJJJJJJJJJ7JAJFFJJJJJJJ7JAFJJJ
$ zcat SRR5837342_isize148_149_r2.fq.gz | grep -A 3 -w 'SRR5837342.10000218'
@SRR5837342.10000218 10000218/2
TGACAGCTGGGCTGCGCTCCGGGCCGCGGCAAGCCGCGAGCGCGCAGGCGCAGCAGCCCCGCGCCGCGCTAGGCAGGCCTGGAGGGCGGGACAAGCGCTAGAAGCTCCCGGCCGGGGGCGCGCACGTAGGCACGCAGAGGCCGTCACGCT
+
AAAFFJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJFJAJFJ<FFFJJJFFF<JJJJJJJJJFFJJJJFJJJFFJJJ7FJAJFJAFFJJJ-<FA<
We could manually align them. The 5’ end of r1 and r2 are reverse complementary to each other (r1 on top):
5'- CGTGACGGCCTCTGCGTGCCTACGTGCGCGCCCCCGGCCGGGAGCTTCTAGCGCTTGTCCCGCCCTCCAGGCCTGCCTAGCGCGGCGCGGGGCTGCTGCGCCTGCGCGCTCGCGGCTTGCCGCGGCCCGGAGCGCAGCCCAGCTGTCACT
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
TCGCACTGCCGGAGACGCACGGATGCACGCGCGGGGGCCGGCCCTCGAAGATCGCGAACAGGGCGGGAGGTCCGGACGGATCGCGCCGCGCCCCGACGACGCGGACGCGCGAGCGCCGAACGGCGCCGGGCCTCGCGTCGGGTCGACAGT -5'
This is the situation we mentioned before. The read length is actually longer than the insert size. In this case, a successful trimming workflow would trim off the extra 2 bp at the 3’ end of both reads. This is indeed the case in the fastp trimmed fastq:
$ zcat fastp_isize148_149_r1.fq.gz | grep -A 3 -w 'SRR5837342.10000218'
@SRR5837342.10000218 10000218/1
CGTGACGGCCTCTGCGTGCCTACGTGCGCGCCCCCGGCCGGGAGCTTCTAGCGCTTGTCCCGCCCTCCAGGCCTGCCTAGCGCGGCGCGGGGCTGCTGCGCCTGCGCGCTCGCGGCTTGCCGCGGCCCGGAGCGCAGCCCAGCTGTCA
+
AAFFFJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJFJJJJJJJJJJJJJJJJJJJ7JAJFFJJJJJJJ7JAFJ
$ zcat fastp_isize148_149_r2.fq.gz | grep -A 3 -w 'SRR5837342.10000218'
@SRR5837342.10000218 10000218/2
TGACAGCTGGGCTGCGCTCCGGGCCGCGGCAAGCCGCGAGCGCGCAGGCGCAGCAGCCCCGCGCCGCGCTAGGCAGGCCTGGAGGGCGGGACAAGCGCTAGAAGCTCCCGGCCGGGGGCGCGCACGTAGGCACGCAGAGGCCGTCACG
+
AAAFFJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJFJAJFJ<FFFJJJFFF<JJJJJJJJJFFJJJJFJJJFFJJJ7FJAJFJAFFJJJ-<F
As expected, fastp successfully trimmed off the extra 2 bp at both ends of the read, making a pair of 148 bp reads that were reverse complementary to each other. However, if we look at the the cutadapt trimmed fastq, the reads are unchanged:
$ zcat cutadapt_isize148_149_r1.fq.gz | grep -A 3 -w 'SRR5837342.10000218'
@SRR5837342.10000218 10000218/1
CGTGACGGCCTCTGCGTGCCTACGTGCGCGCCCCCGGCCGGGAGCTTCTAGCGCTTGTCCCGCCCTCCAGGCCTGCCTAGCGCGGCGCGGGGCTGCTGCGCCTGCGCGCTCGCGGCTTGCCGCGGCCCGGAGCGCAGCCCAGCTGTCACT
+
AAFFFJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJFJJJJJJJJJJJJJJJJJJJ7JAJFFJJJJJJJ7JAFJJJ
$ zcat cutadapt_isize148_149_r2.fq.gz | grep -A 3 -w 'SRR5837342.10000218'
@SRR5837342.10000218 10000218/2
TGACAGCTGGGCTGCGCTCCGGGCCGCGGCAAGCCGCGAGCGCGCAGGCGCAGCAGCCCCGCGCCGCGCTAGGCAGGCCTGGAGGGCGGGACAAGCGCTAGAAGCTCCCGGCCGGGGGCGCGCACGTAGGCACGCAGAGGCCGTCACGCT
+
AAAFFJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJFJAJFJ<FFFJJJFFF<JJJJJJJJJFFJJJJFJJJFFJJJ7FJAJFJAFFJJJ-<FA<
In this case, the extra 2 bp would affect the mapping. Now let’s look at how exactly this read is mapped to the genome by bowtie2:
# from fastp
$ samtools view fastp_sorted.bam | grep -w 'SRR5837342.10000218'
SRR5837342.10000218 163 chr18 9913859 42 148M = 9913859 148 TGACAGCTGGGCTGCGCTCCGGGCCGCGGCAAGCCGCGAGCGCGCAGGCGCAGCAGCCCCGCGCCGCGCTAGGCAGGCCTGGAGGGCGGGACAAGCGCTAGAAGCTCCCGGCCGGGGGCGCGCACGTAGGCACGCAGAGGCCGTCACG AAAFFJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJFJAJFJ<FFFJJJFFF<JJJJJJJJJFFJJJJFJJJFFJJJ7FJAJFJAFFJJJ-<F AS:i:-6 XN:i:0 XM:i:1 XO:i:0 XG:i:0 NM:i:1MD:Z:74C73 YS:i:-6 YT:Z:CP
SRR5837342.10000218 83 chr18 9913859 42 148M = 9913859 -148 TGACAGCTGGGCTGCGCTCCGGGCCGCGGCAAGCCGCGAGCGCGCAGGCGCAGCAGCCCCGCGCCGCGCTAGGCAGGCCTGGAGGGCGGGACAAGCGCTAGAAGCTCCCGGCCGGGGGCGCGCACGTAGGCACGCAGAGGCCGTCACG JFAJ7JJJJJJJFFJAJ7JJJJJJJJJJJJJJJJJJJFJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJFFFAA AS:i:-6 XN:i:0 XM:i:1 XO:i:0 XG:i:0 NM:i:1MD:Z:74C73 YS:i:-6 YT:Z:CP
# from cutadapt
$ samtools view cutadapt_sorted.bam | grep -w 'SRR5837342.10000218'
SRR5837342.10000218 81 chr18 9913857 42 150M = 9913859 152 AGTGACAGCTGGGCTGCGCTCCGGGCCGCGGCAAGCCGCGAGCGCGCAGGCGCAGCAGCCCCGCGCCGCGCTAGGCAGGCCTGGAGGGCGGGACAAGCGCTAGAAGCTCCCGGCCGGGGGCGCGCACGTAGGCACGCAGAGGCCGTCACG JJJFAJ7JJJJJJJFFJAJ7JJJJJJJJJJJJJJJJJJJFJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJFFFAA AS:i:-18 XN:i:0 XM:i:3 XO:i:0XG:i:0 NM:i:3 MD:Z:0C0T74C73 YS:i:-15 YT:Z:DP
SRR5837342.10000218 161 chr18 9913859 42 150M = 9913857 -152 TGACAGCTGGGCTGCGCTCCGGGCCGCGGCAAGCCGCGAGCGCGCAGGCGCAGCAGCCCCGCGCCGCGCTAGGCAGGCCTGGAGGGCGGGACAAGCGCTAGAAGCTCCCGGCCGGGGGCGCGCACGTAGGCACGCAGAGGCCGTCACGCT AAAFFJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJFJAJFJ<FFFJJJFFF<JJJJJJJJJFFJJJJFJJJFFJJJ7FJAJFJAFFJJJ-<FA< AS:i:-15 XN:i:0 XM:i:3 XO:i:0XG:i:0 NM:i:3 MD:Z:74C73T0G0 YS:i:-18 YT:Z:DP
As we can see, the fastp trimmed reads were successfully and correctly mapped to the genome. The cutadapt trimmed reads that are actually untouched original reads were also successfully mapped to the genome in a incorrect way like this:
Ref (5'->3'): CCTTGACAGCTGGGCTGCGCTCCGGGCCGCGGCAAGCCGCGAGCGCGCAGGCGCAGCAGCCCCGCGCCGCGCTAGGCCGGCCTGGAGGGCGGGACAAGCGCTAGAAGCTCCCGGCCGGGGGCGCGCACGTAGGCACGCAGAGGCCGTCACGTG
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||*|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||**
R2 (5'->3'): TGACAGCTGGGCTGCGCTCCGGGCCGCGGCAAGCCGCGAGCGCGCAGGCGCAGCAGCCCCGCGCCGCGCTAGGCAGGCCTGGAGGGCGGGACAAGCGCTAGAAGCTCCCGGCCGGGGGCGCGCACGTAGGCACGCAGAGGCCGTCACGCT
**||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||*|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
R1 (3'<-5'): TCACTGTCGACCCGACGCGAGGCCCGGCGCCGTTCGGCGCTCGCGCGTCCGCGTCGTCGGGGCGCGGCGCGATCCGTCCGGACCTCCCGCCCTGTTCGCGATCTTCGAGGGCCGGCCCCCGCGCGTGCATCCGTGCGTCTCCGGCAGTGC
See that the extra 2 bp overhang at the 3’ end of the reads together with a mismatch in the middle of the reads (a total of three mismatches) are tolerated during bowtie2 mapping, which resulted in a dovetail alignment. This alignment was treated as not properly paired, and it was filtered out by the -f2 flag. The fragment length is also incorrectly set as 152 bp due to the dovetail.
If we manually check some other reads, we realised that all of them are in the situation described above. Now the question becomes: why cutadapt failed to trim the extra 1 or 2 bp at the 3’ end of the reads? It seems to be successfully and correctly trimmed the reads when the overhang is 3 or more bp. This led me to take a closer look at the cutadapt.stdout which I often overlooked. Most of the time, I only look at the % of reads that have the adapters and the % of reads left. However, there is extra information that is useful at the end of the log. For example, this is a few lines of the “Overview of removed sequences” section from the output from SRR5837342:
Overview of removed sequences
length count expect max.err error counts
3 130890 188714.1 0 130890
4 53819 47178.5 0 53819
5 34523 11794.6 0 34523
6 30372 2948.7 0 30372
7 30723 737.2 0 30723
8 30063 184.3 0 30063
9 31567 46.1 0 30842 725
10 33603 11.5 1 32327 1276
As expected, by default, the cutadapt just does not trim adapters that are less than 3 bp. In the case described here, if the insert size is too short, we would expect read 1 and read 2 to be reverse complementary to each other, so we can confidently trim off the adapters even if it is only 1 bp at the 3’ end1. There must be an option to adjust that. It turns out it is the -O --overlap option:
-O MINLENGTH, --overlap MINLENGTH
Require MINLENGTH overlap between read and adapter for an adapter to be found.
Default: 3
Now, let’s try that:
# adapter removal
cutadapt -j 20 -m 25 -O 1\
-a CTGTCTCTTATACACATCTCCGAGCCCACGAGAC \
-A CTGTCTCTTATACACATCTGACGCTGCCGACGA \
-o cutadapt_O1_r1.fq.gz -p cutadapt_O1_r2.fq.gz \
SRR5837342_1.fastq.gz SRR5837342_2.fastq.gz \
1> cutadapt_O1.stdout 2> cutadapt_O1.stderr
# bowtie2 mapping
bowtie2 -p 20 -X 2000 \
--very-sensitive \
-x <hg38_bwt2_idx> \
-1 cutadapt_O1_r1.fq.gz -2 cutadapt_O2_r2.fq.gz | \
samtools view -@ 20 -hu - | \
samtools sort - -@ 20 -T tmp -o cutadapt_O1_sorted.bam
# look at the fragment (isize)
samtools view -F 4 -f 66 -q 30 cutadapt_O1_sorted.bam | \
sed '/chrM/d' | \
awk '{ if($9>0) {print $9} else {print -1*$9} }' | \
sort | uniq -c | sort -b -k2,2n | \
awk 'BEGIN{OFS="\t"}{print $2, $1}' > cutadapt_O1_f2q30_isize.tsv
First, the log is promising, showing the vast majority of the reads got trimmed off by 1 bp:
Overview of removed sequences
length count expect max.err error counts
1 1285763 3019425.2 0 1285763
2 408198 754856.3 0 408198
3 130890 188714.1 0 130890
4 53819 47178.5 0 53819
5 34523 11794.6 0 34523
6 30372 2948.7 0 30372
7 30723 737.2 0 30723
8 30063 184.3 0 30063
9 31567 46.1 0 30842 725
10 33603 11.5 1 32327 1276
Then we check the fragment distribution:
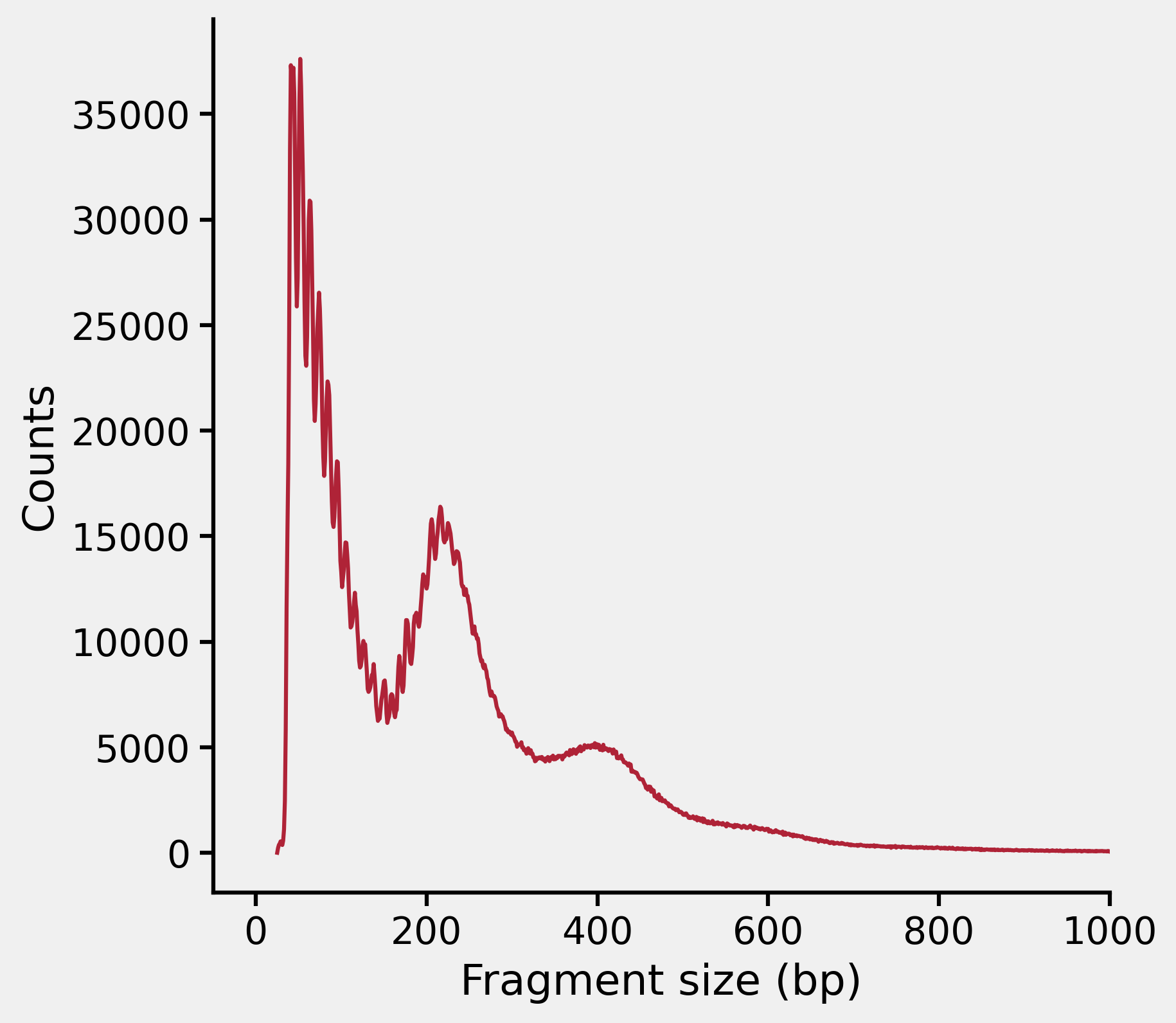
Finally, we have solved the problem. If I really paid attention to all the content in the log file, it would save me a lot of time.