NOTE: this post is just a slightly more detailed version of this Twitter thread.
Not very long after I wrote the series about guessing the library layout of PIP-seq, the raw data (FASTQ files) for the V4 chemistry were made available from the FluentBio website, which is really nice.
Now let’s have a look at what has been changed. The experimental procedures seem to remain the same. What about the library layout and barcodes? Let’s use the T20 Kit V4 Barnyard Study data to figure that out. With the experience from the previous versions, we can immediately start with FastQC of Read 1.
This is the result of SC20230206_FR_1_S1_L001_R1_001.fastq.gz:
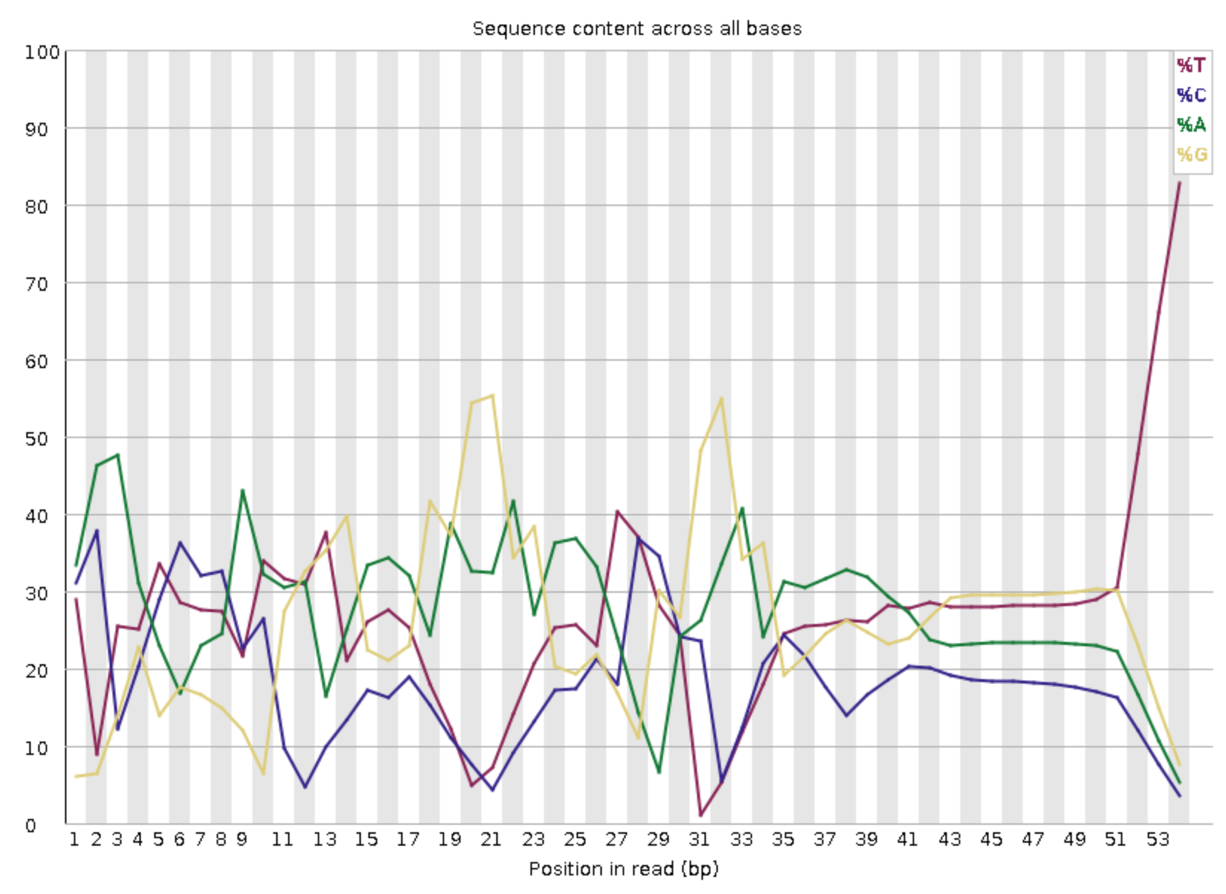
We do not see any common sequences in the middle of the read, like those in the V2 and V3. If we assume that the barcodes are still generated by the split-pool method, it often means there are some variable bases near the beginning part of the read to “de-sync” the sequencing, increasing the base complexity in each sequencing cycle. Another hint can be found in the last few bases of the read. We see that the dominant base is T, so this is basically the oligo-dT. However, unlike the V2 chemistry where almost 100% of the read have T at the last few bases, we only have ~80% of T at most. This also indicates there are some variable bases near the start of the read.
In this case, we need to do some manual alignment using the trick we mentioned previously. Get the first 40 high-quality sequences ignoring reads containing N by:
zcat SC20230206_FR_1_S1_L001_R1_001.fastq.gz | \
sed -n '2~4 p' | \
head -40
Put the output into VS Code. Bear in mind that there should be some common sequences around the middle part of the read, we can start manually aligning them like this:
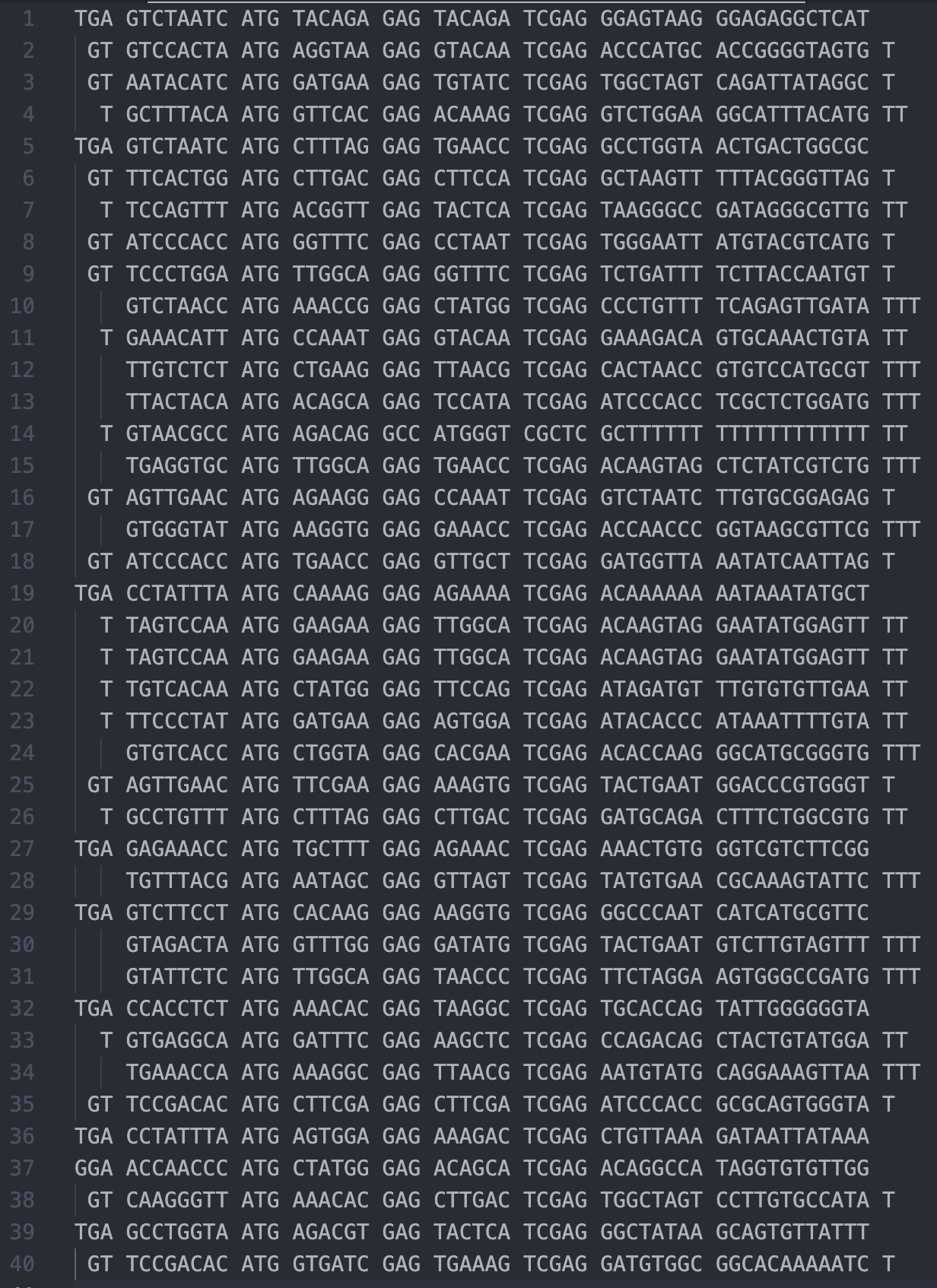
Now it is clear to us the read structure is: None/T/GT/TGA + 8bp bc1 + ATG + 6bp bc2 + CAG + 6bp bc3 + TCGAG + 8bp bc4 + 12 bp UMI + T. That is almost the same as the V3 chemistry, except the variable bases None/T/GT/TGA at the beginning. Therefore, we have reasons to believe that the whitelist remains unchanged.
Is this the case? Let’s find out by processing the data using the V3 whitelist. Since we know the read structure, this can be done by STARsolo using the following command:
STAR --runThreadN 40 \
--genomeDir reference/others/hs_mm_mix/star_2.7.9a \
--readFilesCommand zcat \
--outFileNamePrefix ./star_outs/ \
--readFilesIn SC20230206_FR_1_S1_L001_R2_001.fastq.gz \
SC20230206_FR_1_S1_L001_R1_001.fastq.gz \
--soloType CB_UMI_Complex \
--soloBarcodeReadLength 0 \
--soloAdapterSequence ATGNNNNNNGAGNNNNNNTCGAG \
--soloCBposition 2_-8_2_-1 2_3_2_8 2_12_2_17 2_23_2_30 \
--soloUMIposition 2_31_2_42 \
--soloCBwhitelist fb_v3_bc1.tsv fb_v3_bc2.tsv fb_v3_bc3.tsv fb_v3_bc4.tsv \
--soloCBmatchWLtype 1MM \
--soloCellFilter EmptyDrops_CR \
--outSAMattributes CB UB \
--outSAMtype BAM SortedByCoordinate
Once it is done, we can check the content of the Summary.csv:
Number of Reads,1067049515
Reads With Valid Barcodes,0.953267
Sequencing Saturation,0.397782
Q30 Bases in CB+UMI,0.964693
Q30 Bases in RNA read,0.935793
Reads Mapped to Genome: Unique+Multiple,0.857187
Reads Mapped to Genome: Unique,0.648044
Reads Mapped to Gene: Unique+Multipe Gene,0.719135
Reads Mapped to Gene: Unique Gene,0.692885
Estimated Number of Cells,40579
Unique Reads in Cells Mapped to Gene,677629098
Fraction of Unique Reads in Cells,0.916529
Mean Reads per Cell,16699
Median Reads per Cell,15482
UMIs in Cells,407207180
Mean UMI per Cell,10034
Median UMI per Cell,9348
Mean Gene per Cell,2571
Median Gene per Cell,2581
Total Gene Detected,43477We see that more than 95% of the barcodes are valid, which confirms our guess.
Another sanity check to do is whether the first base of Read 2 is all T. We expect this to be the case since the library preparation procedures are almost the same as V3. The Read 2 adapter is added through fragmentation, A tailing and ligation. Therefore, all Read 2 should start with a T. Now let’s have a look:
$ zcat SC20230206_FR_1_S1_L001_R2_001.fastq.gz | sed -n '2~4 p' | cut -c 1 | sort | uniq -c
286396621 A
129912129 C
264907938 G
385832827 T
It is not what we expected. Maybe the first cycle of Read 2 is a dark cycle. I’m not entirely sure.