This is the third post from a series where I use the recent PIP-seq data as an example to demonstrate the importance to use a common standard like seqspec to accompany sequencing reads submissions into public repositories.
Some Notes
In the previous post, we have figured out the library structure of many samples from the Clark2023 paper. They are using the commerical FluentBio kits, so they basically have the PIPseq$^{\textmd{TM}}$ V3 library structure. However, we still haven’t figure out what is going on with samples like SRR19086119. We will tackle this in the next post.
On top of SRR19086119 and the like, there is still another version of the method. Check the “GSM6138415 Mixed species” sample, which eventually leads to SRR19180490. We can download the FASTQ files:
$ fastq-dump --split-3 -A SRR19180490
PIPseeker$^{\textmd{TM}}$ cannot recognise the FASTQ files, so it is neither v3 nor v4. I’m pretty sure it is not used in the commercial version, but it was used in the Clark2023 paper. Therefore, I call this PIP-seq V2. This is just me giving it a name, not necessarily correct.
SRR19180490
We already had some previous experience, so we could immediately check Read 1 with FastQC:
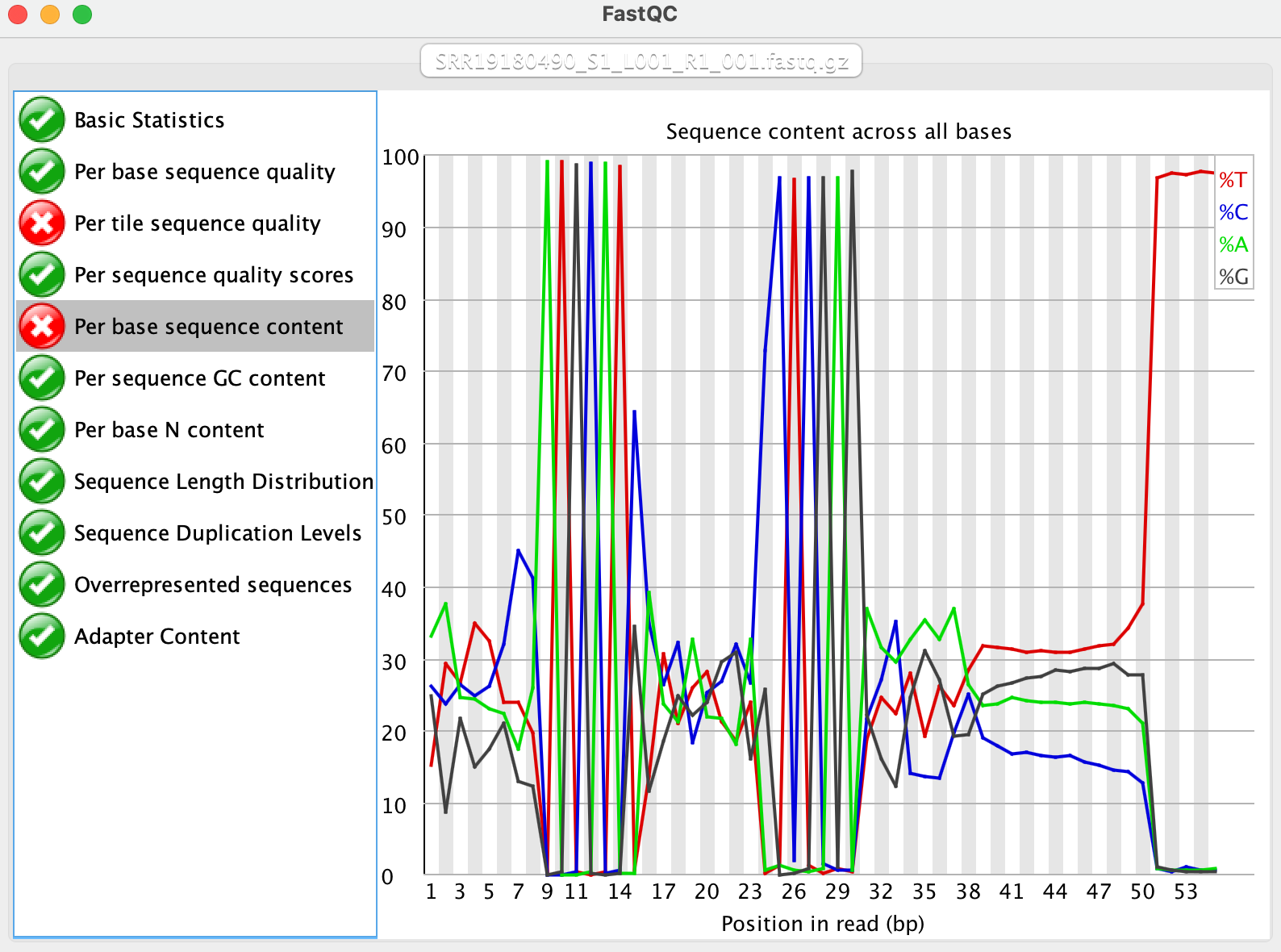
It is immediately clear to us that there are some constant regions (linkers or splints), so it is reasonable to guess the variable regions flanking the linkers are barcodes or UMIs:
| Base positions | Identity |
|---|---|
| 1 - 8 | a variable region (possibly 8 bp barcode1) |
| 9 - 15 | ATGCATS (Here, “S” is ~65% C and ~35% G) |
| 16 - 23 | a variable region (possibly 8 bp barcode2) |
| 24 - 30 | SCTCGAG (Here, “S” is ~75% C and ~25% G) |
| 31 - 38 | a variable region (possibly 8 bp barcode3) |
| 39 - 50 | a variable region (possibly 12 bp UMI) |
| 51 - 55 | TTTTT (this is from oligo-dT) |
In the constant regions, there is a base with S (G or C). Not sure why this happens, but we are going to just use the most frequent base to represent them. That is, ATGCATS is ATGCATC, and SCTCGAG is CCTCGAG. It does not affect the results.
Naturally, we would guess there should be fixed sequences in those three variable regions. Let’s find out using the similar technique from the previous post:
zcat SRR19180490_1.fastq.gz | \
sed -n '2~4 p' | \
cut -c 1-8 | \
sort | uniq -c |
sort -b -k1,1nr | \
less -N
The output is all unique 8-mers in the first 8 bases for Read 1, sorted by occurrences in descending order. As expected, the occurrences of the top 96 8-mers are within a similar range, but there is a sudden an order of magnitude drop from the 96th to the 97th 8-mers. Therefore, we are relatively confident that the top 96 8-mers are in our whitelist. Here are the lines 94 - 98 of the output:
| |
Similarly, we could check bases 16 - 23:
zcat SRR19180490_1.fastq.gz | \
sed -n '2~4 p' | \
cut -c 16-23 | \
sort | uniq -c |
sort -b -k1,1nr | \
less -N
Lines 94 - 102 from the output are shown here:
| |
In this case, the differences of occurrences among 96th - 100th 8-mers are not very different. There is a sudden an order of magnitude drop in frequency from 100th to 101st 8-mer. What’s going on with the 97th - 100th 8-mers? If we check carefully enough, we could find out that each of them has only 1 bp variation compared to some of the 8-mers from the top 96. Therefore, it seems we could still just use the top 96 as the whitelist.
Bases 31 - 38 are in the similar situation. Therefore, we could extract the whitelist by:
# get barcode1 (8 bp)
zcat SRR19180490_1.fastq.gz | \
sed -n '2~4 p' | \
cut -c 1-8 | \
sort | uniq -c | \
sort -b -k1,1nr | \
head -96 | \
awk '{print $2}' > pip-seq_v2_bc1.tsv
# get barcode2 (8 bp)
zcat SRR19180490_1.fastq.gz | \
sed -n '2~4 p' | \
cut -c 16-23 | \
sort | uniq -c | \
sort -b -k1,1nr | \
head -96 | \
awk '{print $2}' > pip-seq_v2_bc2.tsv
# get barcode3 (8 bp)
zcat SRR19180490_1.fastq.gz | \
sed -n '2~4 p' | \
cut -c 31-38 | \
sort | uniq -c | \
sort -b -k1,1nr | \
head -96 | \
awk '{print $2}' > pip-seq_v2_bc3.tsv
On top of that, by checking the method section of the Clark2023 paper, we finally figure out the library structure of PIP-seq V2. The HTML representation is here, and the seqspec specification is here.
Validation
Once we got the whitelist for all three parts, we could start the validation using STARsolo. The command is straightforward to understand after we figured out the library structure:
STAR --runThreadN 40 \
--genomeDir /data/bio-chenx/reference/others/hs_mm_mix/refdata-gex-GRCh38-and-mm10-2020-A/star_2.7.9a \
--readFilesCommand zcat \
--outFileNamePrefix ./star_outs/SRR19180490/ \
--readFilesIn fastqs/SRR19180490_2.fastq.gz fastqs/SRR19180490_1.fastq.gz \
--soloType CB_UMI_Complex \
--soloCBposition 0_0_0_7 0_15_0_22 0_30_0_37 \
--soloUMIposition 0_38_0_49 \
--soloBarcodeReadLength 0 \
--soloCBwhitelist pip-seq_v2_bc1.tsv pip-seq_v2_bc2.tsv pip-seq_v2_bc3.tsv \
--soloCBmatchWLtype 1MM \
--soloCellFilter EmptyDrops_CR \
--outSAMattributes CB UB \
--outSAMtype BAM SortedByCoordinate
Check the result summary:
$ cat star_outs/SRR19180490/Solo.out/Gene/Summary.csv
Number of Reads,129331399
Reads With Valid Barcodes,0.934121
Sequencing Saturation,0.360427
Q30 Bases in CB+UMI,0.958061
Q30 Bases in RNA read,0.943714
Reads Mapped to Genome: Unique+Multiple,0.823944
Reads Mapped to Genome: Unique,0.608917
Reads Mapped to Gene: Unique+Multipe Gene,0.682908
Reads Mapped to Gene: Unique Gene,0.656224
Estimated Number of Cells,2932
Unique Reads in Cells Mapped to Gene,52596524
Fraction of Unique Reads in Cells,0.619727
Mean Reads per Cell,17938
Median Reads per Cell,14059
UMIs in Cells,31166927
Mean UMI per Cell,10629
Median UMI per Cell,8632
Mean Gene per Cell,2362
Median Gene per Cell,2541
Total Gene Detected,33440More than 93% reads have valid barcodes, I guess this is quite acceptable. After this, we should use another sample for an independent validation. At this moment of writing, I only found SRR19180490 had this structure, but I was not searching very hard … Oh well …