Although ATAC-seq has been extensively used in the functional genomics field to study chromatin accessibility, it is still under debate how we should perform peak calling to identify oepn chromatin regions. MACS2 is a very popular peak caller for ChIP-seq, and it has been used for ATAC-seq as well. How should we perform ATAC-seq peak calling using MACS2? That is a recurring question. If you pay attention to the details in those publications that uses ATAC-seq, you might notice that some people uses the --shift -100 --extsize 200 flags during MACS2 peak calling.
For us, we routinely convert paired BAM to simple BED file and use the following flags with MACS2 to call ATAC-seq peaks:
-f BED --keep-dup all --nomodel --shift -100 --extsize 200
Why? Here are the reasons:
People tend to remove duplicates using the Picard tools after read alignment. By default, MACS2 will also remove duplicates in the data. If you use Picard, --keep-dup all will tell MACS2 not to perform duplicate removal. If you don’t use Picard, you can remove that option and let MACS2 do the work. The point here is NOT to perform the same step twice with possibly different standards, i.e. Picard vs MACS2.
Like many other peak callers, MACS2 is designed for ChIP-seq. In ChIP-seq, the reads are flanking the actual binding site of the protein. Therefore, many peak callers either shift or extend reads towards the middle of the fragments to reflect the actual binding site. MACS2 uses extension, that is, the --ext flag.
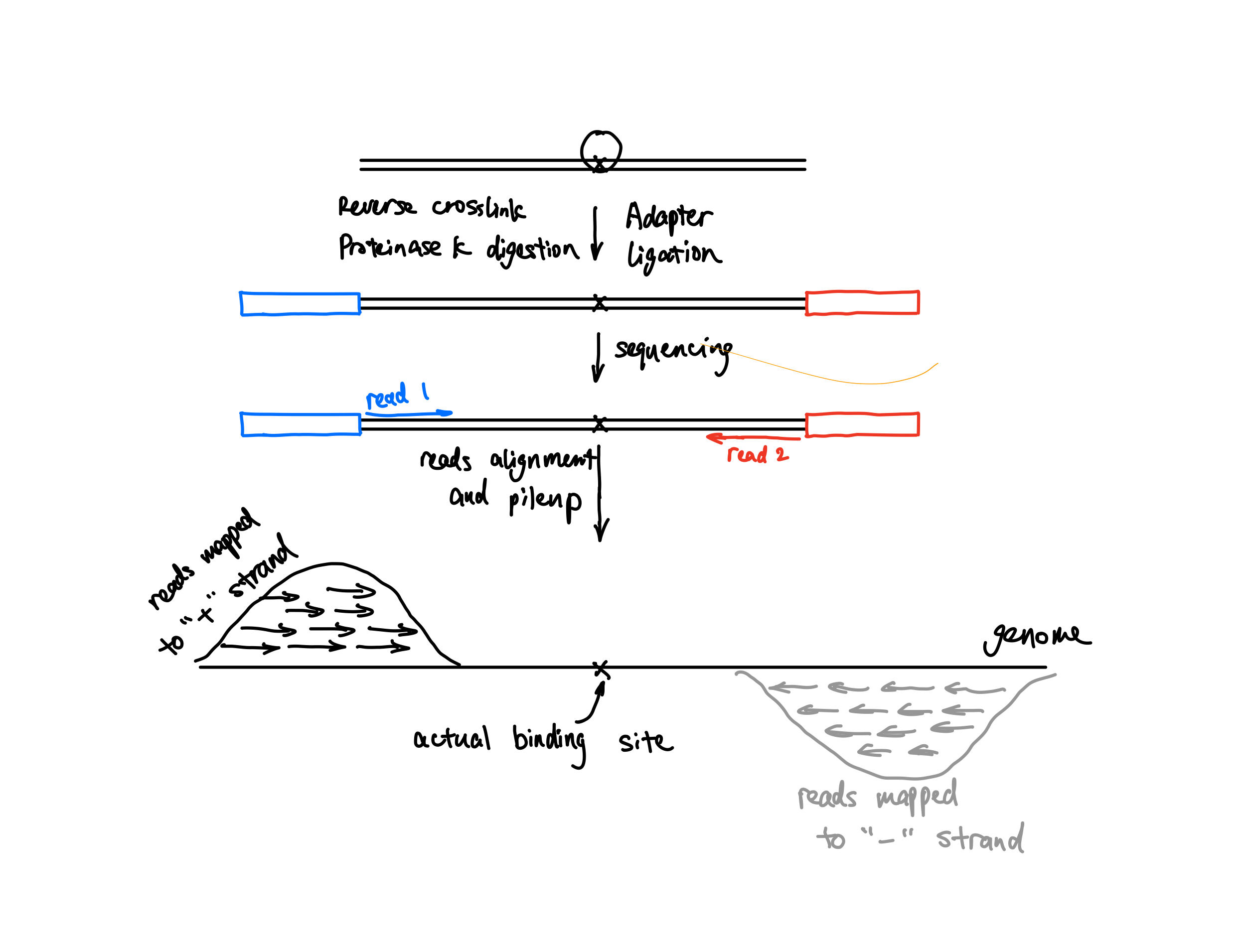
In ATAC-seq/DNase-seq, the middle of the fragments is not really what we are interested in. Instead, we are interested in the blue and red dots in the figure shown below which are the cutting sites of the enzyme. Those dots are the start (5’ end) of your reads, so the default of MACS2 doesn’t fit here.
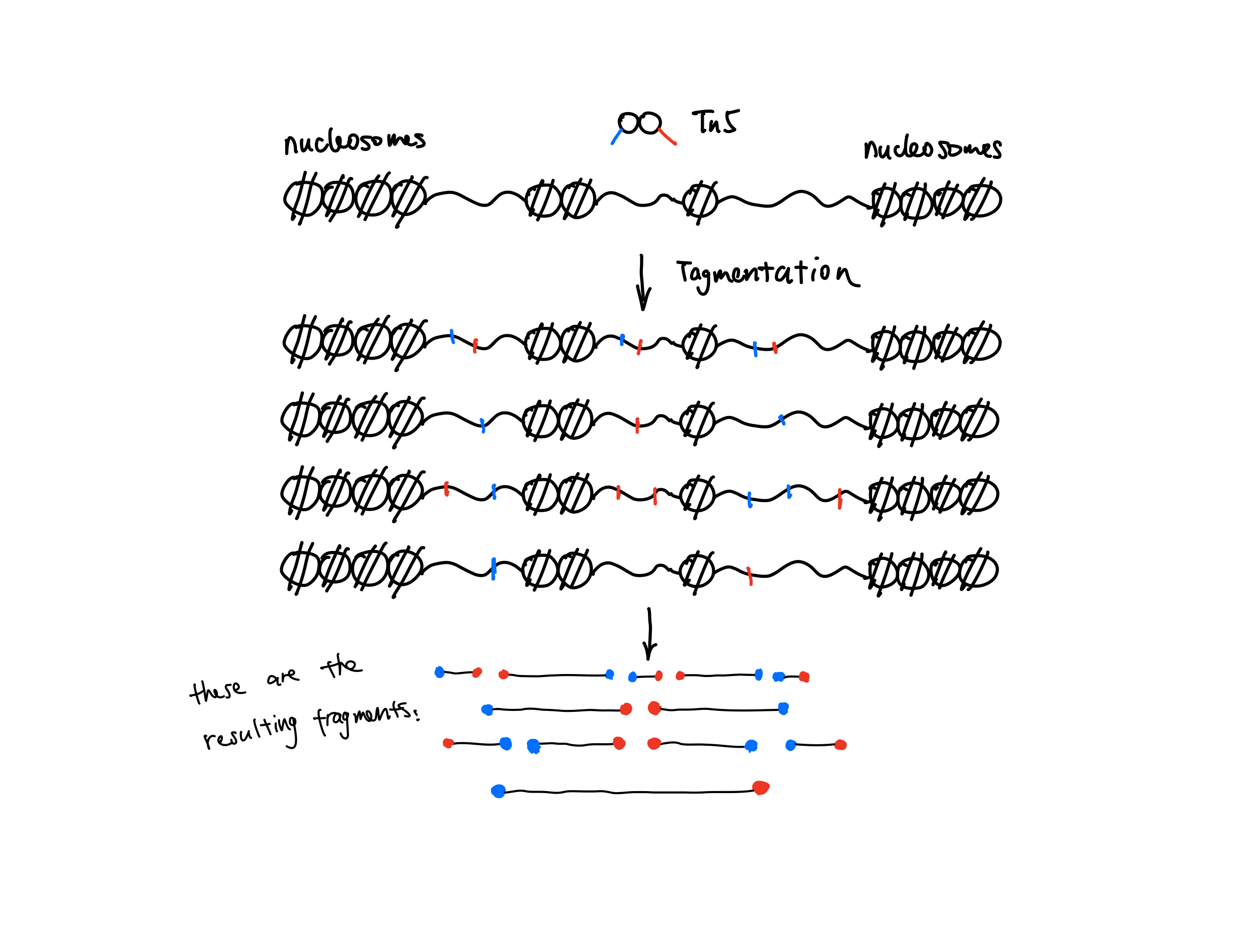
Instead, we want to call peaks with fragments centred on the 5’ end of your reads. We had this discussion about 6 years ago in the MACS2 google usergroup. Both @fooliu and @anshulkundaje provided excellent information. Check this link if haven’t seen it before.
An update of MACS2 (ver 2.1.0 20140616) was made after the discussion, and you could freely manipulate the read positions with the combination of the --shift and the --extsize flags. Of course, when you do that, you need to add the --nomodel flag as well to ask MACS2 not to determine the shift size automatically. Then, why do we covert paired BAM to simple BED to use -f BED?
According to Issue #145 from the MACS2 GitHub, when you set -f BAM or -f BAMPE, MACS2 only takes the left read, ignoring the other. However, in ATAC-seq the 5’ ends of both R1 and R2 are of interest. Convert to BED will solve this problem, I guess?
Finally, why --shift -100 --extsize 200? Well … this is arbituary and it is a habbit from ChIP-seq. To be honest, according to a MNase-seq study (check their Figure 1), it seems that using a fragment size of 75 bp centered on the cutting site might give better resolution. Maybe this chioce is better. In that case, it should be --shift -37 --extsize 75. The ENCODE project used these parameters.
Not sure if all those modifications will make huge difference, but the results are different. I haven’t tried other choices like HMMRATAC, Genrich, MACS3 etc. yet. I will probably do that in future. Hope this helps to those who are new to this type of analysis.
You can also find the Twitter discussion from this thread.